A versatile method for targeting gene replacement in Drosophila was devised in 2000 by Yikang Rong and Kent Golic. We have since used this system to knock out successfully three genes. Suggestions based on our experiences and our discussions with Kent and Yikang are presented below.
Keith Maggert, Wei Gong, and Kent Golic recently published an excellent guide to targeting: Methods for Homologous Recombination in Drosophila. pp 155-174 in Drosophila Methods and Protocols, Christian Dahmann, ed., Humana Press. Additional information (according to me) is that there are more vectors available than those listed (see our vectors page), and you don't need to kinase your oligonucletides when adding an I-SceI (or other type) of site (see below).
Ends-in vs. ends-out Ends-in vs. ends-out
Amount of targeting DNA
Introducing mutations into the gene of interest
Introducing an I-SceI site
Donor insertions
Targeting crosses
Confirming targeting of the homologous gene
Reducing duplications to a single copy
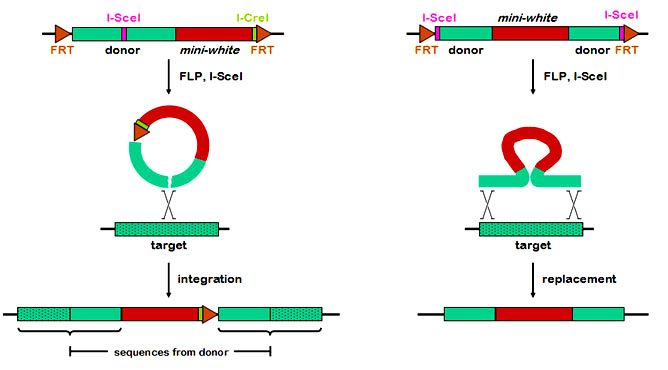
There are two general methods of gene targeting, called ends-in and ends-out. They differ in whether the double-strand break is within the region of homology (ends-in) or at the ends (ends-out). These two methods produce very different products. The figure below compares the two methods in their most basic forms. With ends-in (left), a break is made within the region of homology. Recombination with the target (drawn as a simple crossover, but the actual mechanism is not known) results in a tandem duplication of all the homologous sequence carried on the donor, separated by any additional sequences that are between the FRTs (in this case, the mini-white and I-CreI site. The ends-out method, in contrast, is a simple replacement of the genomic sequence with the homologous sequence. A typical strategy for knocking out a gene might be to interrupt that gene with a heterologous sequence, such as the mini-white marker.
At first glance, the ends-out method seems to be a simpler method of knocking out a gene. The ends-in method, however, may be more versatile. The pTV2 vector of Rong and Golic and the vectors we've built contain an ICreI site, which can be used to "reduce" the duplication back to a single copy without the intervening DNA. This makes it possible to not only knock out the target gene, but to introduce site-directed mutations, in-frame fusions of tags, etc. In addition, it may be that ends-in is more efficient than ends-out, although ends-out seems to work reasonably well in Drosophila.
We've developed a number of useful vectors and constructs for both ends-in and end-out targeting.
Amount of targeting DNA
The relationship between the amount of targeting DNA and the efficiency of targeting has not been systematically studied. The current recommendation is that about 6 kb of genomic sequence be used - targeting efficiency decreases with less than this amount, but does not greatly increase with more than this. The distribution of sequence (left and right of the cut site (ends-in) or insertion (ends-out) has also not been studied, so we recommend a roughly symmetrical distribution.
Genomic DNA sequence can include the gene of interest only, part of the gene of interest, or the gene of interest and several surrounding genes. It is okay to end within a flanking gene. If the construct includes flanking genes and is generated by PCR, however, a polymerase with proof-reading activity should be used to avoid introduction of unwanted mutations into flanking genes.
Introducing mutations into the gene of interest
Ends-out targeting is generally used for destroying gene function. The mutation is usually an insertion of a marker gene
Introducing an I-SceI site
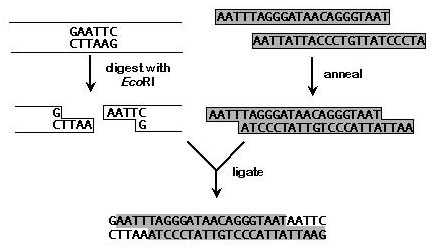
For ends-in targeting, an I-SceI recognition site must be introduced into the genomic DNA on the donor construct. We find it easiest to purchase two oligonucleotides that are complementary except for overhanging ends, anneal them, and ligate them into a unique restriction site with then genomic DNA. Because oligos generally come without 5' phosphates, one needn't worry about concatamerization of the insert. On the other hand, it means the donor construct cannot be treated with phosphatase, and therefore it can easily re-ligate without an insert. To counteract this, we generally use a 10 to 50-fold molar excess of annealed oligonucleotides. In addition, the oligonucleotides can be designed so that the restriction site is destroyed. One can then eliminate re-ligated products by cutting the ligation reaction with the original enzyme prior to transformation. Re-ligation products will be cut, but products with the I-SceI insertion will not. The efficiency of transformation with circular DNA is several orders of magnitude higher than that of linear DNA. An example is shown below.
Donor insertions
Donor insertions are obtained by standard P element germline transformation. For unknown reasons, some insertions work much better than others. Proximity to the target does not seem to be a major factor, nor does being on the same chromosome. For example, in targeting RecQ5, which maps to 70E, we tried insertions in 70D and 71D, but neither worked well. An insertion on 2 worked the best. Because it is as yet impossible to predict whether a particular insertion will work well for targeting, we recommend using as many different insertions as possible. FLP seems to work efficiently on most insertions, but occasionally an insertion is refractory to FLP activity. It is easy to cross each line to the constitutive FLP source to test this; lines that do not FLP out readily (as monitored by eye color mosaicism) should not be used.
Targeting crosses
To induce targeting, donor construct lines are crossed to the P{hsp70::FLP} P{hsp70::I-SceI} stock (see example below). The progeny are subjected to heat shock to induce FLP and I-SceI. We have heat-shocked on days 3-5, with little difference in efficiency. Heat shocking should be done by placing vials into a water bath at 38 C, for 90 minutes. One or two broods of 10-20 vials per donor construct should be sufficient.
Virgin females with the donor and the FLP and I-SceI constructs are collected when they eclose. These females should have white eyes, possibly with a few colored cells remaining, indicating FLP activity. These are crossed to males that constitutively express FLP (as in the example below), and the progeny are screened for any with solid eye color.
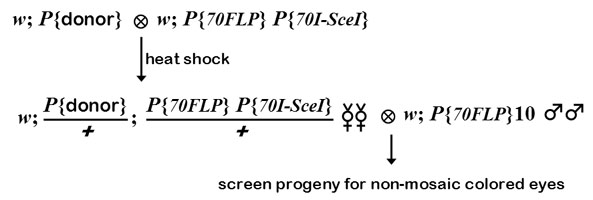
In this example, a donor construct on 2 is crossed to a stock with FLP and I-SceI constructs on 3. It is possible to do the targeting without keeping track of all these constructs in the progeny - you can replace chromosomes later to get of unwanted stragglers. The first cross must be done in vials to get good heat exchange during the heat shock, but the second cross can be done in bottles. It is possible to use males in this step, but in our experience and that of the Golic lab, targeting is more efficient in the female germline. Potential targeting events will be recognized as progeny with colored eyes. The largest sources of non-targeted events with colored eyes are (1) cases in which the donor never excised, and (2) cases in which unequal FRT recombination between FRTs on sister chromatids resulted in a tandem duplication of the donor (we found this to be especially prevalent in the male germline). Rong and Golic came up with a clever way to eliminate these background events from further consideration. In both cases mentioned, there are FRTs surrounding the mini-whitegene, and thus FLP can excise mini-white. An insertion of excised targeting DNA, however, will have one (for ends-in) or no (for ends-out) FRTs remaining, and therefore the mini-white gene will not be excised by FLP. This is why we cross to the constitutive FLP stock: solid colored eyes indicates a potential targeting event, whereas mosaic eyes indicates an undesired event (the vast majority of progeny will have white eyes). Rong and Golic have made available P{hsp70::FLP}10, which is a homozygous-viable insertion on 2 that expresses FLP constitutively, without heat shock. One should be aware that targeting may (and probably does) occur pre-meiotically. Therefore, it is possible to obtain clusters of progeny with colored eyes in the same bottle. These may or may not represent identical events.
Confirming targeting of the homologous gene
The first step in confirming targeting is to map the mini-white marker to a chromosome. If it maps to the chromosome of the target gene, there are several possible methods to distinguish insertion/replacement of the homologous gene from random insertion. These include genetic mapping, Southern blotting, and PCR. Genetic mapping can give you an idea of whether the mini-white is in the right neighborhood, but it takes a month. An example is when we knocked out the mus81 gene (actually, Steve Brill's lab built the construct, and Kim McKim's lab obtained donor inserts and potential targeting events, before sending them to us). this gene maps to base of the X chromosome, near yellow. We therefore mapped three potential targeting events relative to y. One was far from y, and two were tightly linked, and were therefore considered excellent candidates.
Southern blotting is the most straightforward method for determining not only whether targeting has occurred as desired, but to confirm that the event has the predicted structure. We were fooled by a Southern blot of a putative end-in event at Ercc1. A deletion was created during the targeting event, such that the band we predicted to be larger came out the same size as wild-type.
PCR is the fastest method for confirming targeting. However, since most donor constructs will carry 3 kb of DNA to either side of the I-SceI site (for ends-in) or the mini-white insertion (for ends-out), it may be difficult to amplify the diagnostic product. We have found inverse PCR to be a viable alternative. One can cut with an enzyme for which there is only a single site in the donor construct (e.g., the XbaI within the FRT or the NcoI within mini-white), and for which there is a known site in the flanking DNA at the target locus. Note that this requires ligation of a larger circle (>3 kb) than usual, and this may necessitate changes to the standard inverse PCR protocols.
Reducing duplications to a single copy
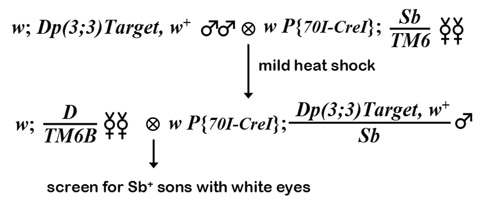
The ends-in method of targeting generated a tandem duplication of the region contained on the construct, with vector sequences (mini-white and the I-CreI site) separating the two units. Reducing this to a single copy is accomplished by crossing to a source of the I-CreI endonuclease, then selecting for loss of mini-white activity. The targeted duplication is crossed to P{hsp70::I-CreI}, and the progeny are subjected to heat-shock. Rong and Golic believe that this enzyme can cut Drosophila rDNA, so the recommend a mild heat shock - 36 C for 1 hour. Single male progeny with the tandem duplication and I-CreI construct are crossed to an appropriate balancer stock. Their progeny (usually sons) are screened for white eyes. These are then crossed to an appropriate stock, and checked for those that retain a single copy of the target, carrying the desired mutation.
|
|
|
|
|
|
|
© Newtron Laboratories






